Sequencing RNA
Which RNA molecules do you want to sequence?
mRNA:
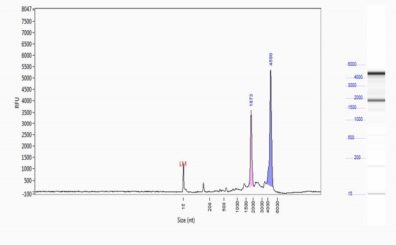
90-98% of all the RNA purified from a cell will be ribosomal RNA (rRNA) that takes the form of several discreet molecular species involved in translating proteins from messenger RNA (mRNA). The mRNA is regularly used as a proxy for expressed genes but must be purified away from other RNA species before sequencing. Enrichment can be accomplished either by affinity purification of the mRNA using the polyA tail to hybrdize to a polyT oligonucleotide on a matrix (oligo dT purification) or by selective removal of rRNA by attaching rRNA hybridization probes to a matrix (ribo depletion).
Oligo dT purification is wonderfully selective for mature mRNA but lncRNA, smRNA, micoRNAs, circRNAs, etc. will be lost from the sample. Ribo-depletion can be used if you are interested in investigating these other interesting forms of RNA in addtion to the mRNA.
Other RNAs:
smRNA (40-200bases) and miRNA (10-40bases) which are highly regulatory, require special library construction techniques.
lncRNA is investigated after removal of both rRNA and mRNA and size selecting away miRNA, smRNA and tRNAs.
Microbial mRNA in host: Requires both prokaryotic and eukaryotic probes for rRNA depletion and removal of host polyA mRNA.
Circular RNA: Resesarch suggests most transcripts have circular forms. These enodnuclease resistant molecules are more persistent, appear to have many regulatory functions, and may help explain some of the discrepancies between the transcriptome and the proteome. PolyA purification will cause the loss of circRNA. In ribo-depleted samples the software CircExplorer can be use to indentify the circularizing reads. If you are only intersted in circRNA, endonuclease treatment is used to remove linear molecules before library construction.
What do you want to learn?
Expression values of know genes:
In RNA-Seq, sequencing reads are mapped onto a reference genome allowing simple counting of fragments mapped onto know features to generate relative abundance of each feature in the sample. RNA-seq displaced hybridization based micro-arrays by overcoming the detection threshold and saturation limit. Simply counting generates digital data without saturation. All cells have genes whose expression is barely detectable as well as many that are wildly abundant. For vertebrate sized genomes we target 20million reads/sample. If you are interested in the rarest of the rare short lived regulatory transcripts, you will need more sequence. If you are interested in abundant proteins, that is far more sequence than you need. All of our RNA-seq is performed on the Ion Proton using a P1 semiconductor sequencing chip which can generate 60-80 million reads twice a day. For high sample numbers or single cell RNA-seq libraries using the Chromium X system, an Illumina sequencing instrument is the prefered platform. The Ion Proton is best for modest sample number, fast turn-around, and methods development.
Human and Mouse AmpliSeq transcriptomes are expression panels that provide a faster, cheaper method to generate expression values than RNA-Seq for these two organisms, using much less starting material. Ampli-seq is the sequencing of a massively multiplexed amplicon pool on our Ion Proton. In addition to cancer gene and other diagnostic panels, panels are available that target 22k expressed features in the mouse or human genome. Since these libraries are amplified we can start with as little as 50pg of total RNA and still generate a reproducible set of mRNA expression values. Like a micro-array, there is no discovery of anything that isn’t targeted on the panel. Features of most interest are included allowing Ampli-Seq to replace RNA-Seq for many researchers. Fewer reads are needed/sample in ampliseq since we are mapping everything to 22k short expected amplicons. With ampliseq transcriptomes we target 5-10million reads/sample. Amplicon targets can be found in the .bed file for each panel. <Ampliseq Transcriptome Mouse.bed> <Ampliseq_Transcriptome_Human.bed>
Transcript Discovery:
For researchers without a model genome looking for a special transcript, denovo assembly of the RNA sequence into transcripts and functional annotation is performed in place of mapping to a reference
Full Length Transcript Structure:
Iso-Seq is used to profile full length mRNA using a PacBio sequencer. Most genes have multiple transcripts that differ in the 5` transcription initiation site, 3′ transcription termination site, and contain variable internal splicing. RNA transcripts reconstructed from short reads often fail to correctly define the various mRNA transcripts for a given gene. With long reads now available, higher quality transcript models are possible. Isoform sequencing (Iso-Seq) with the PacBio provides a clearer window into a transcriptome’s isoform variants. The process can not be used to count relative abundance, only provide a transcript profile.
Something new:
New RNA species and methods are always being reported. We are always willing to help you develop or use a new method.